Análisis de secuencias del COVID-19
Resumen
Acabo de leer una reciente publicación en una de las revistas científicas más prestigiosas de ciencia (PNAS) sobre los genomas y las mutaciones encontradas hasta ahora en un pequeño grupo analizado.
Es decir que se ha analizado la secuencia de bases del virus y las mutaciones entre distintos casos de diferentes lugares
Cito parte del estudio traducido: «En un análisis de red filogenética de 160 genomas completos del síndrome respiratorio agudo grave humano 2 (SARS-Cov-2), encontramos tres variantes centrales distinguidas por cambios de aminoácidos, que hemos denominado A, B y C, siendo A el tipo ancestral según el coronavirus del grupo de salida de murciélagos. Los tipos A y C se encuentran en proporciones significativas fuera de Asia oriental, es decir, en europeos y estadounidenses. Por el contrario, el tipo B es el tipo más común en Asia oriental, y su genoma ancestral parece no haberse extendido fuera de Asia oriental sin antes mutar en tipos B derivados, apuntando a efectos fundadores o resistencia inmunológica o ambiental contra este tipo fuera de Asia. La red rastrea fielmente las rutas de infecciones por casos documentados de enfermedad coronavirus 2019 (COVID-19), lo que indica que las redes filogenéticas también pueden utilizarse con éxito para ayudar a rastrear fuentes de infección COVID-19 no documentadas, que luego pueden ponerse en cuarentena para prevenir la propagación recurrente de la enfermedad en todo el mundo.
Las secuencias de nucleótidos de los genomas sars-CoV-2 utilizados en este análisis están disponibles, previa inscripción gratuita, en la base de datos GISAID (https://www.gisaid.org/). El paquete de software Network5011 y los archivos de red coronavirus están disponibles como shareware en el sitio web de Fluxus Technology (https://www.fluxus-engineering.com/).
Análisis de la red filogenética de los genomas SARS-CoV-2
Peter Forster, Lucy Forster, Colin Renfrew, Michael Forster
Actas de la Academia Nacional de Ciencias Abril 2020, 202004999; DOI: 10.1073/pnas.2004999117
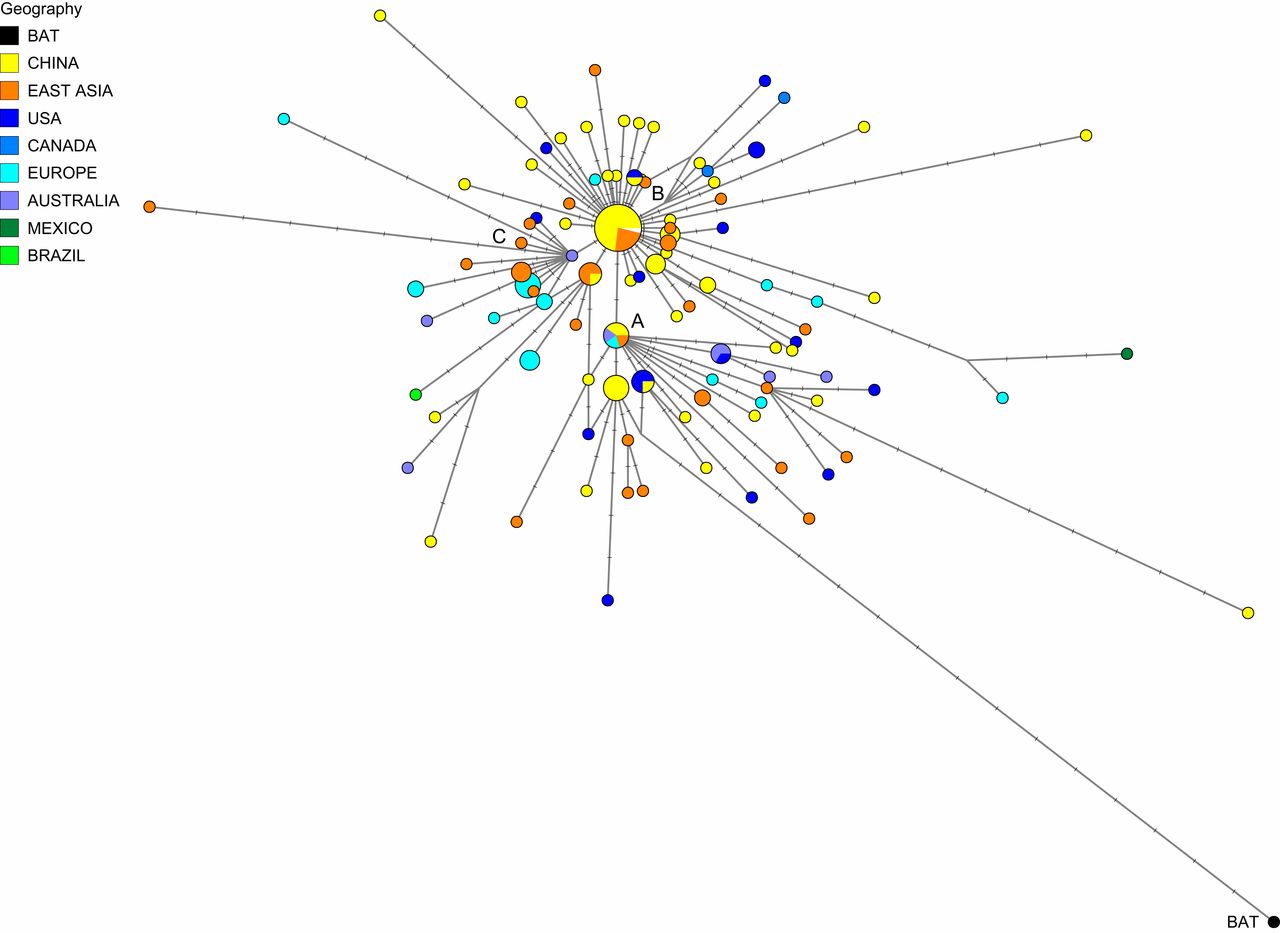
Red filogenética de 160 genomas SARS-CoV-2. El nodo A es el cúmulo raíz obtenido con el murciélago (R. affinis) aislado del coronavirus BatCoVRaTG13 de la provincia de Yunnan. Las áreas circulares son proporcionales al número de taxones, y cada muesca en los enlaces representa una posición de nucleótido mutado. El rango de secuencia considerado es de 56 a 29.797, con numeración de posición de nucleótido (np) según la secuencia de referencia Wuhan 1 (8). Se utilizaron el algoritmo de red de unión a la mediana (2) y el algoritmo Steiner (9), ambos implementados en el paquete de software Network5011CS(https://www.fluxus-engineering.com/),con el parámetro epsilon establecido en cero, generando esta red que contiene 288 árboles más parsimoniosos de 229 mutaciones de longitud 229. Las reticulaciones son causadas principalmente por mutaciones recurrentes en np11083. Los 161 taxones (160 virus humanos y un virus de murciélago) producen 101 secuencias genómicas distintas. El diagrama filogenético está disponible para el escrutinio detallado en formato de póster A0(Apéndice SI, Fig. S5) y en la red gratuita archivos de des
Les dejo la imágen del estudio comparativo de las secuencias
Espero les parezca interesante e informativo como a mi.
Para leer el articulo completo les dejo enlace aquí