Hoy quería dejarles la monografía realizada por Mariana de Gregorio acerca de la Talasemia. Espero esto les sirva a todos los que buscan información del tema
Trabajo final de
Genética básica.
TALASEMIA.
Autora:
Mariana De Gregorio Paolasini.
Profesora: Gabriela Iglesias.
Fecha: 3 de Noviembre del año 2015.
Universidad: Nacional de Rio Negro.
Curso: Genetica básica, Tercer año.
Carrera: Medicina veterinaria
Introducción:
En la sangre encontramos distintos tipos celulares, entre ellos, los eritrocitos, los cuales representan el número más abundantes de células de la sangre, y que tienen como componente principal la hemoglobina , cuya función es transportar el oxígeno hacia los diferentes tejidos del cuerpo.
Participa en el proceso por el que la sangre lleva los nutrientes necesarios hasta las células del organismo y conduce sus productos de desecho hasta los órganos excretores. También transporta el oxígeno desde los pulmones (o desde las branquias, en los peces), donde la sangre lo capta, hasta los tejidos del cuerpo.
Los eritrocitos son producidos continuamente en la médula ósea de los huesos largos principalmente. Tienen una forma oval, bicóncava, aplanada, con una depresión en el centro; diseño óptimo para el intercambio de oxígeno con el medio, ya que le otorga flexibilidad para poder atravesar los capilares, donde liberan la carga de oxígeno.
El diámetro de un eritrocito típico es de 6-8 µm.
Los valores considerados normales de eritrocitos en adultos son:
- Mujeres: 4 – 5 x 106/mL(mililitro) de sangre
- Hombres: 4,5 – 5,5 x 106/mL(mililitro) de sangre. wikipedia.org,. (2015).
Un déficit o disminución por debajo del rango de referencia de los eritrocitos genera un estado patológico denominado anemia, cuya alteración provoca hipoxia tisular. En cambio, un exceso de estos, se denomina policitemia, el aumento de la concentración de eritrocitos (eritrocitosis) es una patología mucho menos común.
Existen alteraciones en la maduración de los eritrocitos, entre las cuales están la deficiencia de hierro y las anomalías genéticas que conducen a la producción de hemoglobinas anormales.
Entre las patologías que se pueden producir por anomalías genéticas esta la talasemia, trastorno sanguíneo hereditario.
En este trabajo se explicara que es la talasemia, se nombraran sus variedades, haciendo hincapié en una en particular, llamada β-talasemia, dentro de la que encontramos más de 200 tipos de mutaciones, de las que se explicaran las más frecuentes en nuestro país.
Desarrollo:
En un sujeto normal, los glóbulos rojos tienen una duración de 120 días de vida. Cada día, el cuerpo produce nuevos glóbulos rojos para reemplazar los que han muerto o los que el cuerpo ha perdido. En la talasemia, los glóbulos rojos se destruyen a una velocidad mayor generando anemia.
La talasemia ¨Es un trastorno sanguíneo que se transmite de padres a hijos (hereditario) en el cual el cuerpo produce una forma anormal de hemoglobina, la proteína en los glóbulos rojos que transporta el oxígeno¨. (Policlinicalacibis.es,. 2015).
Esta hemoglobina está compuesta por cuatro cadenas de polipeptidos, dos cadenas de globina alfa y dos cadenas de globina beta. Por lo que hay dos tipos de talasemia principales – talasemia alfa y talasemia beta – cuyo nombre viene de los defectos que pueden ocurrir en estas cadenas de proteínas.
Hay dos copias del gen que produce la hemoglobina α (HBA1 y HBA2), y cada uno codifica una cadena α, y ambos genes están localizados en el cromosoma 16. El gen que codifica las cadenas β (HBB) está localizado en el cromosoma 11. (Es.wikipedia.org,. 2015).
Lo que genera las siguientes patologías:
- Alfa talasemia:cuando el cuerpo tiene dificultades produciendo alfa globina
- Beta talasemia:cuando el cuerpo tiene dificultades produciendo beta globina
α-talasemia
El gen HBA1 y HBA2, del cromosoma 16, hay una deficiencia de síntesis de cadenas α. El resultado es un exceso de cadenas β que trasportan deficientemente el oxígeno, lo que conduce a bajas concentraciones de O2 (hipoxemia).
β-talasemia
Hay una falta de cadenas β, y el consiguiente exceso de cadenas alfa, que puede formar agregados insolubles que se adhieren a la membrana de los eritrocitos, pudiendo causar la muerte de éstos y sus precursores, originando anemia de tipo hemolítico.
Estas compensaciones dan lugar a la formación de hemoglobinas inestables que provocan la destrucción de los glóbulos rojos y por lo tanto anemia. (Es.wikipedia.org,. 2015).
La talasemia se transmite de manera autosómica recesiva, afectando a los varones y mujeres igualmente, pues no implica el cromosoma de sexo y se da cuando existe un defecto en un gen que ayuda a controlar la síntesis de una de las proteínas globulina alfa o globulina beta que componen la hemoglobina.
Como se explico anteriormente, hay diversas formas de talasemia y cada tipo tiene muchos subtipos diferentes. Tanto la talasemia α como la talasemia β, abarcan las siguientes dos formas, dependiendo la severidad de los síntomas:
- Talasemia menor.
- Talasemia mayor.
1. Talasemia Menor
La talasemia menor se presenta si uno recibe el gen defectuoso de solo uno de los padres. Las personas con esta forma de trastorno son portadoras de la enfermedad y por lo general no tienen síntomas.
2. Talasemia mayor: Es necesario heredar el gen defectuoso de ambos padres para padecer la talasemia mayor.
Esta enfermedad está provocada por deleciones en uno o varios genes de los que componen los grupos de la α-globina y la β-globina. Según la cantidad de deleciones, el tipo de talasemia será más o menos grave.
Existen otras deleciones como resultado de entrecruzamientos desequilibrados entre los segmentos duplicados presentes en la región de la agrupación. (Es.wikipedia.org,. 2015).
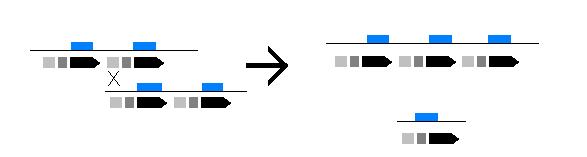
Imagen de entrecruzamiento desequilibrado o desigual de cromátides. Upload.wikimedia.org,. (2015)
Talasemia alfa:
La talasemia alfa ocurre cuando un gen, o los dos genes relacionados con la proteína globina α de la hemoglobina faltan o se han modificado, mutado.
La alfa globina se genera en el cromosoma 16, por lo tanto, si los dos genes que le indican al cromosoma 16 que produzca alfa globina no se encuentran o han mutado, se produce menos alfa globina.
Esto afecta la hemoglobina y disminuye la capacidad de los glóbulos rojos de transportar oxígeno por el cuerpo.
“Se necesitan cuatro genes, dos de cada padre, para hacer cadenas de proteína alfa. Cuando faltan uno o más de los genes, se produce la talasemia alfa. Este gráfico describe los diferentes tipos de talasemia.”
| Genes alfa que faltan | Problema | Síntomas de anemia | Otros nombres |
| 1 | Portador silencioso | Ninguno | Talasemia alfa – 2 rasgos, talasemia alfa mínima |
| 2 | Rasgos | Leve | Talasemia alfa – 1 rasgo, talasemia alfa menor |
| 3 | Hemoglobina H | Moderados | Enfermedad de la hemoglobina H |
| 4 | Seria | Mortal | Hidropesía fetal con la Hemoglobina de Bart |
Clinica de Cleaveland consultado en 2015.

- Portador silencioso de alfa talasemia: un alelo del gen de la cadena alfa está delecionado (los otros tres son normales). Genotipo -/α α/α
- Portador de alfa talasemia:perdida de dos alelos α, de los genes de cadena alfa, cualquiera ambos del mismo cromosoma 16, llamaron una canceladura de los «cis» o uno de ambos cromosomas 16, llamado una canceladura «trans.» Genotipo: -/- α/α or -/α -/α.
- Enfermedad de la hemoglobina H: perdida de tres alelos α de los dos genes de la cadena alfa están delecionados. La enfermedad de la hemoglobina H, produce una anemia. Las personas que tienen la enfermedad de la hemoglobina H corren un mayor riesgo de tener un hijo con alfa talasemia grave, puesto que son portadores de un cromosoma número 16 con dos genes delecionados de la cadena alfa (deleción en cis). Genotipo: -/- -/α
- Alfa talasemia grave:pérdida de los cuatro alelos α, de ambos genes de la cadena alfa, lo que es tan grave que puede producirse la muerte dentro del útero (antes del nacimiento). Genotipo: -/- -/-
- Todos los casos posibles de talasemia alfa, según la ausencia de uno, dos, tres o cuatro genes de la alfa globina. Es.wikipedia.org, (2015).
Ventajas de la talasemia α:
La α-talasemia protege a los individuos que la portan frente a la malaria. La malaria o paludismo está producida por un parásito protista del género Plasmodium y es transmitida por un mosquito del género Anopheles. La protección frente a esta enfermedad por parte de los individuos que posee α-talasemia es debida a que Plasmodium sólo es capaz de parasitar a los eritrocitos sanos.
Sin embargo, la sangre de alguien con este tipo de anemia presenta un número elevado de eritrocitos deformes por culpa de que la hemoglobina no está bien constituida y eso es esencial pues deja al parásito indefenso en la sangre permitiendo que nuestro sistema inmunitario acabe con él.
Talasemia Beta
Normalmente hay dos genes de globina beta, uno heredado de cada padre. La talasemia beta es un cambio en uno o los dos genes de globina beta, localiza en el cromosoma 11. Las mutaciones pueden suprimir completamente (mutaciones β0) o disminuir (mutaciones β+ y β++) la producción de cadenas β globina, lo que resulta en un desequilibrio en la síntesis de cadenas de globina α/β.
La magnitud de este, es la determinante principal del fenotipo de la enfermedad, que abarca desde los individuos asintomáticos (β-Talasemia menor o portador) que agrupa a los genotipos heterocigotos (β+/P o β0/P) y que corresponde a la forma más frecuente en nuestro país, hasta los que dependen de transfusiones regulares para vivir (β talasemia mayor) que comprende a los genotipos homocigotos (β0/β0 y β+/β+) o dobles heterocigotos (β0/β+), y corresponde a las formas de mayor expresividad clínica.
Entre ambos extremos, se encuentran los pacientes con β talasemia intermedia (BTI), en los cuales las manifestaciones clínicas son variables.
Este gráfico describe los diferentes tipos de talasemia beta.
| Genes beta afectados | Problema | Síntomas de anemia | Otros nombres |
| 1 | Portador silencioso | Leve | |
| 1 | Rasgo | Leve | |
| 2 | Intermedia | Moderado | |
| 2 | Mayor | Severo | Anemia de Cooley |
Clinica de Cleaveland consultado en 2015.
También existen casos de deleciones de diversos tamaños que pueden afectar al gen de la beta globina o a la región de control del locus.
Mayoritariamente es una enfermedad hereditaria con un patrón autosómico recesivo, pero también existen algunos casos donde la herencia es autosómica dominante.
También existen dos variedades de beta-talasemia (mayor o menor) según sea un déficit total o parcial de la síntesis (dependiendo la severidad de los síntomas): la talasemia mayor (también conocida como anemia de Cooley o anemia del mediterráneo) que es más severa y la talasemia intermedia.
–Beta talasemia grave o MAYOR u homocigota (anemia de Cooley): los dos genes de la cadena beta tienen deleciones, causando el tipo más grave de beta talasemia. Los pacientes que tienen talasemia grave pueden fabricar suficientes glóbulos rojos por lo que necesitan frecuentes transfusiones de sangre y puede que no vivan mucho tiempo.
Durante el primer año o dos primeros años de vida, pueden estar pálidos, irritables, tener poco apetito y padecer muchas infecciones. Sin tratamiento, aumenta el tamaño del hígado, del bazo y del corazón, y los huesos pueden volverse delgados y quebradizos, desarrollan hemosiderosis (depósito en todos los tejidos del hierro liberado tras la hemólisis).
Es frecuente la presencia de cálculos biliares por la hemólisis crónica. Adquieren un color pardo-verdoso por la anemia, la ictericia (la hemólisis libera bilirrubina que produce un color amarillo en la piel y mucosas) y la hemosiderosis. Se detiene el crecimiento, se retrasa la pubertad. Y finalmente se produce un fallo cardíaco.
Actualmente algunos pacientes pueden también ser tratados, e incluso curados, mediante un transplante de médula ósea.
Beta talasemia leve o característica de talasemia – un gen beta tiene una deleción, provocando anemia. La talasemia leve se divide en:
1.-Talasemia mínima (la persona tiene pocos o ningún síntoma).
2.-Talasemia intermedia (la persona tiene una anemia de moderada a grave).
-Beta Talasemia Intermedia: Se designa así al síndrome talasémico de moderada intensidad, que condiciona la aparición de una anemia leve y alteraciones óseas.
Presentan sintomatología clínica y requieren transfusiones de sangre durante alguna época de su vida, pueden desarrollar hemosiderosis.
Sus manifestaciones no son tan graves como en los pacientes afectados de la forma mayor de la enfermedad.
-Beta talasemia heterocigota o menor (rasgo talasemico): aparece cuando sólo está afectada una de las copias del gen que codifica la cadena. Es la mutación del gen beta, caracterizada por unos hematíes elevada, con concentración de hemoglobina normal o disminuida y generalmente presenta un aumento de la Hb A2.
Las personas portadoras de talasemia menor, no presentan manifestaciones clínicas, aunque en ocasiones pueden tener una ligera anemia que se pone de manifiesto al realizar un análisis.
Los glóbulos rojos de los portadores del rasgo talasémico son más pequeños de lo normal. La talasemia menor está presente desde el nacimiento, permanece durante toda la vida y puede transmitirse de los padres a los hijos.
Las β-talasemias además de la deleción del gen de la β-globina, también pueden darse por otras causas como:
- Mutaciones en el promotor que detienen o reducen su transcripción.
- Mutaciones en los sitios de corte y empalme (splicing) que impiden la eliminación de los intrones.
- Mutaciones en el sitio aceptor de poli-A que afectan al procesamiento del mesnajero ó mRNA.
- Mutaciones de cambio en la pauta de lectura.
Es.wikipedia.org,. (2015).
O también pueden presentarse otras formas de talasemia beta cuando se hereda un gen para la beta talasemia en combinación con un gen de una variante hemoglobínica. Las más importantes son:
- HbE: Si se hereda un gen de la HbE y uno de la beta talasemia, esta combinación es la responsable de la HbE-beta talasemia, apareciendo una anemia de moderada a severa similar a la beta talasemia intermedia.
- HbS: beta talasemiao anemia falciforme. Si se hereda un gen de la HbS y otro de la beta talasemia, aparece la HbS-beta talasemia. es,. (2015).
Diagnostico para un paciente talasemico:
El diagnostico se puede realizar con una única muestra de sangre, realizando:
- Cuadro Hemático Completo (CBC), que incluye la medición de la hemoglobina y la cantidad/ tamaño de células rojas. La gente que sufre de talasemia tiene menos cantidad de células rojas sanas, menos hemoglobina de lo normal y dichos eritrocitos serán más pequeños e irregulares. (hemograma completo).
- Un recuento de reticulocitos (medición de células rojas jóvenes) puede indicar que tu médula espinal no está produciendo el número adecuado de células rojas.
- Los estudios del hierro indicarán si la causa de la anemia es una deficiencia de hierro (anemia ferropenica) o talasemia.
- Se pueden usar pruebas genéticas o análisis mutacional para diagnosticar cuando hay un historial familiar de talasemia.
- Electroforesis de la hemoglobina: es unprocedimiento de laboratorio que diferencia los tipos de hemoglobina presentes.
- El médico lleva a cabo un examen físico para buscar un bazo inflamado (agrandado).
DIAGNOSTICO MOLECULAR- PCR:
La PCR es un método sencillo para la amplificación in vitro de cualquier segmento de ADN permitiendo disponer de forma rápida, eficaz y económica, de cantidades suficientes del mismo para su posterior estudio molecular. Mediante esta técnica, se realiza la detección de los genotipos causantes de β-talasemia, ya que permiten discriminar entre alelos normales y mutantes que difieren en una sola base.
El método Amplificación Refractaria de Sistemas de Mutaciones (ARMS-PCR) es una modificación de la técnica de PCR, utilizada para la detección de mutaciones puntuales causantes de β-talasemia.
Esta técnica permite la amplificación enzimática de alelos específicos, mediante el uso de cebadores que están diseñados para discriminar entre secuencias que difieren en una única base. Además, utiliza cebadores control que amplifican otra región del gen de β globina, cercana a la mutación que será detectada, actuando como control interno de amplificación asegurando la eficiencia de la PCR y evitando falsos negativos.
Este es un método basado en la reacción en cadena de la polimerasa, capaz de detectar diversas mutaciones puntuales y pequeñas deleciones o inserciones en el gen β globina, con el empleo de oligonucleótidos (primers o cebadores)de secuencia específica.
Los productos obtenidos en la amplificación (PCR) pueden analizarse mediante diversas técnicas:
- Dot blot: se utiliza para detección de mutaciones puntuales mediante muestras de ADN hibridadas con sondas marcadas radiactivamente específicas de ciertas regiones del ADN de estudio. Solamente las muestras portadoras de la región de interés se revelan (puntos oscuros).
- Visualización del producto en geles de agarosa o poliacrilamida: se utiliza para separar los fragmentos de DNA basado en su tamaño.
- Análisis con enzimas de restricción: Las enzimas de restricción o endonucleasas, son enzimas que cortan los enlaces fosfodiester del material genético a partir de una secuencia que reconocen. Las mismas permiten cortar DNA de hebra doble, donde reconocen secuencias palindrómicas (secuencias que se leen igual en ambas direcciones).
- Secuenciación directa del ADN amplificado: determinación del orden de los nucleótidos (A, C, G y T) en un oligonucleótido de ADN
Las mutaciones más frecuentes en la población argentina del gen de β-globina son CD39 e IVS1-110, las cuales se dan a conocer mediante la técnica de ARMS-PCR.
- IVS-I-110 (G>A)
- CD 39 (C>T),
El diagnóstico molecular de β-talasemia, puede ser útil para brindar el asesoramiento genético entre parejas portadoras. La técnica de ARMS-PCR, reúne los requisitos necesarios de los métodos de diagnóstico: alta especificidad, reproducibilidad y bajo costo.
Por lo tanto constituye un método eficaz para el diagnóstico de β-talasemia en pacientes sin posibilidad de estudio familiar, debido a la falta de uno de los padres.
Esta técnica no sólo permite detectar la mutación causante del padecimiento, sino también determinar si se encuentran en estado homocigoto o heterocigoto.
Los pacientes portadores de éstas (hetrerocigotos β/ βCD39 y β/βIVS1-110) y otras mutaciones β-talasémicas, son generalmente (salvo complicaciones) asintomáticos. Sin embargo, en estado homocigótico producen cuadros clínicos de mayor gravedad (Anemia de Cooley).
De aquí la importancia de detectar a los individuos portadores, y en especial, aquellas parejas con probabilidades de concebir hijos con talasemia mayor, teniendo en cuenta las complicaciones que conlleva la herencia de dicha condición. (Qbpatologica.files.wordpress.com,. 2015).
Tratamiento:
Los tratamientos estándar para los pacientes con talasemias serias son las transfusiones de sangre, quelación de hierro, extirpación del bazo, dosis diarias de ácido fólico, posible extirpación quirúrgica de la vesícula biliar, y trasplante de médula.
- Las transfusiones de sangre cada 4 meses en los pacientes con talasemias moderadas o severas, y cada 2 a 4 semanas para los pacientes con talasemia seria beta. Se pueden necesitar transfusiones ocasionales para la enfermedad de la hemoglobina H o la talasemia intermedia beta.
- La quelación del hierro: extirpación del exceso de hierro del cuerpo. Uno de los riesgos de las transfusiones de sangre es que pueden causar una sobrecarga de hierro, que a su vez puede causar enfermedades del corazón.
- Esplenectomía(extirpación del bazo)
- El trasplante de la médula espinal
- Terapia génica: para lograr que un gen normal se inserte en un genoma del individuo con dicha enfermedad hereditaria.
Factores de riesgo:
- Etnicidad afroamericana, asiática, china o mediterránea.
- Antecedentes familiares del trastorno.
Incidencia
Las talasemias alfa ocurren con mayor frecuencia en personas del sudeste asiático, Medio Oriente, China y en aquellas de ascendencia africana. Las talasemias beta ocurren en personas de origen mediterráneo, y en menor grado, los chinos, otros asiáticos y afroamericanos.
Conclusión:
Luego de llevar a cabo la presente monografía, se pude considerar que la talasemia es una enfermedad muy distribuida por el mundo, muy poco conocida por la población, aunque con una gran incidencia.
Estando al tanto de la gran distribución mundial de esta enfermedad, y la gran variedad de formas en las que se puede presentar, es importante dar a conocer la misma, y que sean detectados y alertados por sus médicos aquellos individuos portadores, en especial, aquellas parejas con probabilidades de concebir hijos con talasemia mayor, teniendo en cuenta las complicaciones que conlleva la herencia de dicha condición.
Bibliografía:
- Acosta, K., Riera, M., Labandera, N., Zapata, P. and Galeano, Z. (2015).Estudio de las mutaciones CD39 e IVS1-110 causantes de beta talasemia mediante ARMS-PCR. [online] Available at: [Accessed 15 Nov. 2015].
- Acosta, K. (2015). [online] Available at: [Accessed 15 Nov. 2015].
· Bragos, M. (2015). Diagnostico molecular: aplicaciones en hemoglobina.. [online] Available at: [Accessed 15 Nov. 2015].
- Brandon, N., Aguirre, M. and Gimenez, C. (2015).HEMOGLOBINA. [online] Available at: [Accessed 15 Nov. 2015].
· Clevelandclinic.org,. (2015). Las Talasemias. Retrieved 14 November 2015, from
· Es.wikipedia.org,. (2015). Eritrocito. Retrieved 15 November 2015, from
- wikipedia.org,. (2015).Talasemia. Retrieved 15 November 2015, from
· Exactas-unam.dyndns.org,. (2015). Retrieved 16 November 2015, from
· Fundatal.files.wordpress.com,. (2015). Retrieved 15 November 2015, from
Google.com.ar,. (2015). Resultado de imágenes de Google para
- org,. (2015). Hemoglobinas Anormales :: FUNDREPA. Retrieved 13 November 2015, from
- org, (2015). Talasemias :: FUNDREPA. [online] Available at: [Accessed 15 Nov. 2015].
- Wikipedia
- Harteveld, C. and Higgs, D. PMC (2010). α-thalassaemia.Orphanet Journal of Rare Diseases, 5(1), p.13.
- com,. (2015).Epidemiología y fisiopatología de la talasemia | Iron Health Alliance. Retrieved 14 November 2015, from
- Imagen de entrecruzamiento desequilibrado. Upload.wikimedia.org,. (2015). Retrieved 15 November 2015, from
- es,. (2015).Labtest – Talasemia. Retrieved 15 November 2015, from
- es,. (2015). Retrieved 14 November 2015, from
{kind=link}
· Qbpatologica.files.wordpress.com,. (2015). Retrieved 15 November 2015, from
- Raposo Gonzalez, R. (2015). [online] Available at: [Accessed 15 Nov. 2015].
· Scielo.org.ar,. (2015). Retrieved 15 November 2015, from
- Torres, A. (2015). Beta talasemia intermedia: características clínicas y estudio molecular. Serie de casos clínicos.Arch Argent Pediat, 113(5). [Accessed 15 Nov. 2015].
- edu.pe, (2015).Talasemia. [online] Available at: [Accessed 15 Nov. 2015].